Balancing Act
Feinberg scientists are investigating how regulating inflammation can be a pathway to treating a range of diseases.
by Melissa Rohman
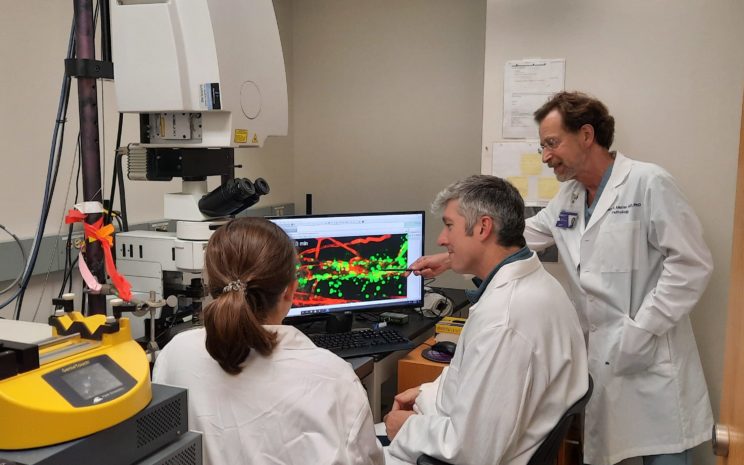
The Muller lab uses four-dimensional intravital confocal microscopy to study transendothelial migration at high resolution in real time in living animals. Here, Muller (far right), graduate student Maureen Hayes (left), and David Sullivan, PhD, research associate professor of Pathology, review data from a recent experiment.
Not all inflammation is created equal. While acute inflammation has evolved as the immune system’s first response to protect the body, chronic inflammation can cause extensive and potentially irreversible damage, as commonly seen in autoimmune diseases or chronic inflammatory diseases like inflammatory bowel disease and atherosclerosis. Regulating inflammation — ensuring that the body doesn’t generate too much of a good thing — is a flourishing area of study at Northwestern.
Perhaps one of the biggest hubs of activity in this area is the lab of William Muller, MD, PhD, the Janardan K. Reddy, MD Professor of Pathology. Muller has been studying the cellular and molecular mechanisms of the body’s inflammatory response in the hope of discovering a more selective approach to treat disease for more than three decades. His lab’s inflammatory models include atherosclerosis, myocardial infarction, dermatitis, and more.
“Simply put, inflammation is at the root of all pathology,” he argues. “Once I realized that chronic inflammation becomes the disease instead of the way to eliminate the disease, I thought it was essential to learn how to regulate inflammation.”
While Muller has studied a broad scope of diseases over the course of his long career, investigators across specialties are chasing the same culprit, studying its effects in diseases ranging from cardiovascular disease and multiple sclerosis (MS) to rheumatoid arthritis and Alzheimer’s disease. Recent high-impact publications demonstrate just how extensively Feinberg scientists have probed this critical factor in disease — and the translational potential their findings hold.
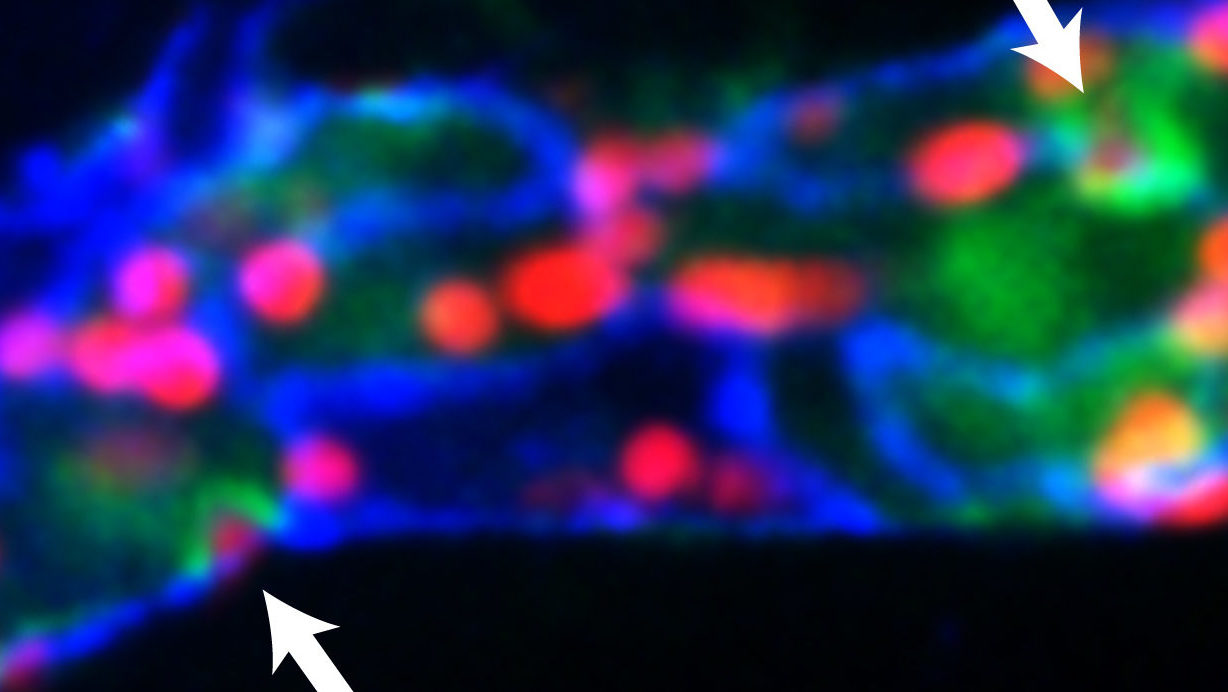
Muller’s lab recently demonstrated that calcium flux in endothelial cells is tightly coupled temporally and spatially with leukocyte transmigration. The arrows depict local elevation of [Ca2+]i (green) at endothelial junctions (blue) as neutrophils (red) transmigrate across a postcapillary venule in the mouse cremaster muscle in response to the inflammatory cytokine, IL-1β.
THE ‘POINT OF NO RETURN‘
The inflammatory response is a sequential process. When tissue is damaged, specialized immune cells already deployed throughout the body detect certain chemicals associated with invasive pathogens as well as damaged or dead cells. These immune cells then dispatch white blood cells, or leukocytes, and other molecular mediators to destroy any remaining pathogens, repair damage, and restore the body’s homeostasis.
Eventually, leukocytes will interact with endothelial cells and move from the blood into the tissue through a process called transendothelial migration, or diapedesis. The phases leading up to diapedesis are reversible. Diapedesis and the phases that follow are not.
Diapedesis, which Muller refers to as the “point of no return,” is therefore an attractive therapeutic target for regulating inflammation and potentially treating chronic inflammatory diseases. Regulating inflammation, however, is a balancing act: The goal is to treat chronic inflammation without eliminating a patient’s entire inflammatory response and rendering them immunocompromised, according to Muller.
“This is the balance we’re trying to strike now: How do we treat someone’s arthritic fingers or their inflammatory bowel disease or their atherosclerosis without rendering them incapable of fighting off infections or healing skin wounds,” Muller says.
In a recent study published in the Journal of Experimental Medicine, Muller’s laboratory discovered that during transmigration, there is a transient increase in endothelial cell calcium ions that are spatially and temporally restricted to the site of diapedesis. The role of the calcium is to activate two key calcium signaling proteins: endothelial calmodulin (CaM) and endothelial calcium/calmodulin kinase II Delta (CaMKII Delta). CaMKII Delta activity is essential for endothelial membrane movement that promotes the transmigration process.
Current anti-inflammatory therapies work by blocking inflammation entirely throughout the body, commonly causing harmful side effects for patients. The findings from the calcium signaling study provide the impetus for regulating inflammation by selectively targeting endothelial cells to decrease the number of white blood cells passing across the endothelial cell wall without interfering with other immune functions, according to Muller. Additionally, his team is beginning to investigate whether diapedesis is regulated similarly across all organs and tissues, specifically in the lungs and brain.
LINKS TO ABNORMAL HEART FUNCTION
One part of the body where inflammation wreaks the most havoc is in the heart. Heart failure is one of the leading causes of morbidity and mortality in the United States. A significant contributor to permanent heart damage after heart attack is due to excessive inflammation caused by reperfusion therapy (a treatment to restore blood flow) that patients receive at the hospital.
A recent study, published in the Journal of Clinical Investigation and led by Edward Thorp, PhD, associate professor of Pathology in the Division of Experimental Pathology, and Matthew DeBerge, PhD, research assistant professor of Pathology, found that an inflammatory cellular pathway previously thought to protect the heart after a heart attack actually causes damage. The team also found that inhibiting this pathway with small molecule inhibitors reduced myocardial infarction-induced damage in mouse models, demonstrating it as a potentially effective therapeutic strategy for patients after a heart attack.
This is the balance we’re trying to strike now: How do we treat someone’s arthritic fingers or their inflammatory bowel disease or their atherosclerosis without rendering them incapable of fighting off infections or healing skin wounds?
William Muller, MD, PHD
Another study, led by Sanjiv Shah, ’00 MD, the Neil J. Stone, MD, Professor of Cardiology, and published in Circulation, found that the presence of inflammatory proteins in the blood was associated with comorbidity burden and abnormal heart function in patients with heart failure with preserved ejection fraction (HFpEF).
“Instead of the heart being the main area of injury, it is inflammation in the bloodstream that appears to be poisoning the heart and the blood vessels in HFpEF,” Shah says.
The findings suggest these proteins could serve as therapeutic targets for patients with HFpEF and act as biomarkers for preventively identifying inflammation.
Ultrastructure of myelinated axons in the central nervous system. Image courtesy of the Popko Laboratory.
FACTOR IN NEUROLOGICAL DISEASES
Neurodegenerative diseases such as Alzheimer’s disease, Parkinson’s disease, and multiple sclerosis (MS), all share a common factor: chronic inflammation.
Inflammation’s role in Alzheimer’s disease has been a focus of Robert Vassar, PhD, the Davee Professor of Alzheimer Research. He was co-author of a study published in Nature that identified a missing link between inflammation and protein deposits that contribute to the development of the disease.
“These findings shed new light on the role of inflammation in Alzheimer’s disease, providing a plausible pathway as well as a potential biomarker or therapeutic target for the condition,” he says.
In the study, investigators conducted an unbiased screen to identify proteins that interact with the gamma-secretase complex, which generates the Abeta protein that accumulates in the notorious amyloid plaques seen in Alzheimer’s disease. That screening identified interferon-induced transmembrane protein 3 (IFITM3).
In mouse models of Alzheimer’s disease and tissue samples from patients with late-stage Alzheimer’s, the investigators found levels of IFITM3 was significantly increased. Deleting IFITM3, however, reduced the production of Abeta. Simulating inflammation also led to increased expression of IFITM3 and subsequently higher levels of Abeta.
“Our study suggests that anti-inflammatory drugs that cross the blood-brain barrier may lower IFITM3 levels and reduce Abeta production, which should delay the onset and progression of Alzheimer’s disease,” Vassar says.
Brian Popko, PhD, the William Frederick Windle Professor of Neurology, has investigated the cellular defense response to inflammation in MS, which affects almost three million people worldwide. Published in the journal eLife, his most recent work suggests that prolonging a cellular defense response to inflammation could help regenerate the protective coating of axons called myelin, which is degraded in diseases like MS.
“This small molecule could be a potential therapeutic because not only have we shown that it provides protection to oligodendrocytes against inflammation, but we now are showing that it promotes myelin repair in an inflammatory environment,” says Popko, who is also scientific director of the Division of Multiple Sclerosis and Neuroimmunology in the Ken and Ruth Davee Department of Neurology. Popko’s team used genetic and pharmacological methods to prolong the integrated stress response in inflammatory models of MS, and discovered that the remyelinating oligodendrocytes, the cells that generate myelin, were protected from inflammation during the repair process. There are currently no FDA-approved drugs that effectively regenerate myelin. The findings, the authors argue, represent a breakthrough therapeutic strategy for treating MS.
A POTENTIAL TARGET FOR RHEUMATOID ARTHRITIS
A team of scientists led by Richard Pope, MD, the Solovy/Arthritis Research Society Professor and professor of Medicine in the Division of Rheumatology, and Deborah Winter, PhD, assistant professor of Medicine in the Division of Rheumatology, discovered that a type of immune cell called tissue-resident macrophage is necessary for the suppression of chronic inflammation.
“This study shows tissue-resident macrophages being the guardian of preventing inflammation and provides a novel way of thinking about treating patients with targeted therapy,” Pope says.
Previous work from Pope’s laboratory found that a specialized protein called FLIP (FLICE-like inhibitory protein) protects macrophages from apoptosis, or cell death, and are also known to be highly expressed in macrophages of patients with rheumatoid arthritis. Using this knowledge, the investigators deleted FLIP from tissue-resident macrophages in mouse models of rheumatoid arthritis to ameliorate the disease. However, deleting FLIP actually promoted chronic inflammation in the mice by preventing monocytes — white blood cells produced in bone marrow — from differentiating into tissue-resident macrophages and performing anti-inflammatory functions.
The findings, which were published in Science Advances, disprove a long-standing notion that all macrophages are “bad” and only promote inflammation.
“If we want to treat people or prevent disease, we can’t just get rid of all macrophages,” Winter says. “We want to promote macrophages that are good for the joints and possibly reprogram the ones that aren’t good.”
As Muller points out, regulating inflammation to treat disease is a balancing act, and scientists across disciplines continue to get closer to answers through shared knowledge.
“It is exciting to perform these studies at a place like Northwestern, where the research culture is truly collaborative. It leads to great opportunities to combine expertise and explore new disease models,” Muller says.
RHEUMATOID ARTHRITIS MEETS PRECISION MEDICINE

Treatment for rheumatoid arthritis has largely been trial and error, but Northwestern scientists are bringing precision medicine to the table.
“I believe this could be game-changing,” says Harris Perlman, PhD, chief of Rheumatology and the Mabel Greene Myers Professor of Medicine, who along with Pope and Winter, was senior co-author of a multisite study that used genetic profiling of joint tissue to see which drugs will work for which patients, published in Arthritis & Rheumatology.
In this study, Perlman and colleagues segregated patients based on the genes being produced by their macrophages, the garbage collectors of the immune system that are overactive in rheumatoid arthritis. They identified two patient groups who shared aspects of the genetic profiles and then which of them showed joint improvement, along with what biologic therapies they were taking. They also identified a gene sequence associated with patients with early disease.
The next goal is to predict which patients will have the best response — based on their genetic signature — to a specific drug.