Untangling Alzheimer’s
by WILL DOSS
From the earliest changes in brain chemistry to novel approaches for clinical care, Northwestern scientists investigate a model spectrum of this devastating disease.

In November 1906, during the 37th Meeting of South-West German Psychiatrists in southern Germany, a physician named Alois Alzheimer described the case of a 50-year-old woman whom he had followed from her admission for paranoia and memory disturbances, until her death five years later.
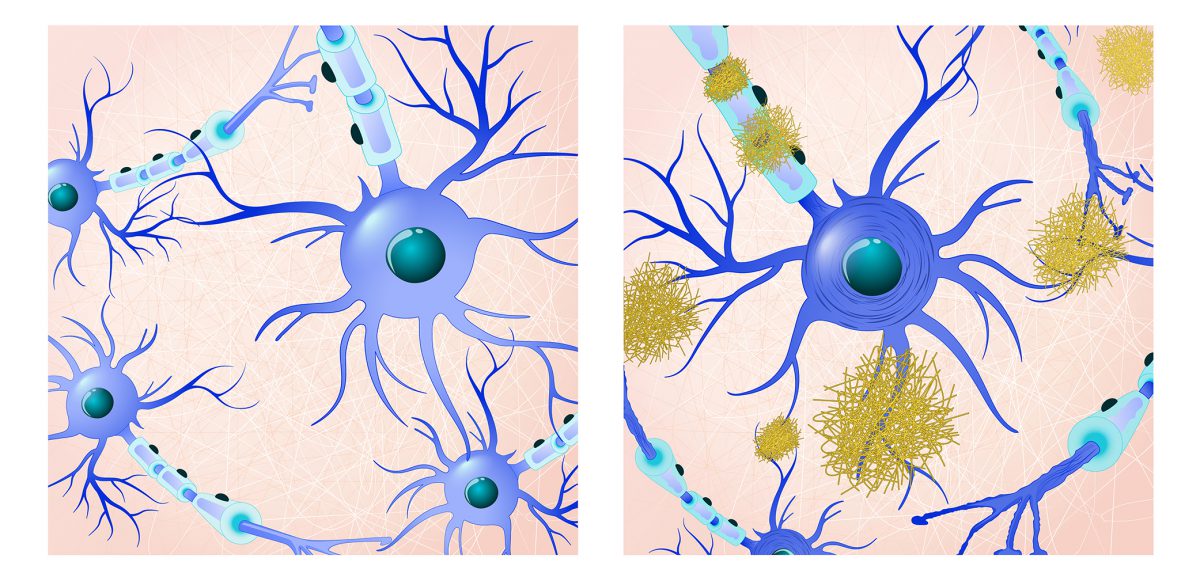
Alzheimer noted “a peculiar severe disease process of the cerebral cortex,” describing distinctive plaque and neurofibrillary tangles in the brain. These characteristics defined the condition later named Alzheimer’s disease.
While the root cause of the disease was largely unknown for decades, scientists discovered the plaques were made of an abnormal protein fragment called amyloid beta, and the theory of the “amyloid cascade” eventually crystallized: Accumulation of amyloid plaques in the brain leads to tangles, which cause an inflammatory response, and eventually, brain cell destruction.
Focusing on this theory, scientists searched for drugs that could slow or stop amyloid beta production. While certain drugs worked well in mouse models of Alzheimer’s disease, it was a different story in humans. Again and again, clinical trials of drugs targeting amyloid beta production ended in failure — and now, the field is at an inflection point, says Jeffrey Savas, PhD, assistant professor in the Ken and Ruth Davee Department of Neurology.

A comprehensive multidisciplinary program, such as the one at the Mesulam Center, derives its strength by making heterogeneity a focus. By appreciating heterogeneity, we can individualize patient care and research into disease mechanisms.
M. Marsel Mesulam, MD
“Decades of work and billions of dollars have been thrown at this amyloid cascade,” Savas says. “People are starting to rethink their assumptions and open up to new ideas.”
While many scientists still believe amyloid plaque accumulation is a major component of Alzheimer’s disease pathology, they increasingly recognize these plaques are not the foundational cause. Furthermore, finding the genesis of Alzheimer’s is compounded by its diversity: the disease comes in several forms, each affected to varying degrees by different disease mechanisms.
To that end, scientists at Northwestern’s Mesulam Center for Cognitive Neurology and Alzheimer’s Disease are investigating a spectrum model of the disease, their inquiry stretching from the earliest changes in brain chemistry to improving diagnosis and treatment.
“A comprehensive multidisciplinary program, such as the one at the Mesulam Center, derives its strength by making heterogeneity a focus of research, not a nuisance to be avoided,” says M. Marsel Mesulam, MD, director of the Mesulam Center. “It is only by appreciating heterogeneity that we can individualize patient care and research into disease mechanisms.”
The Mesulam Center is set up precisely for this cutting-edge approach. “The same room where a patient management conference has just been held becomes the venue of a basic science laboratory meeting, often with a considerable overlap of attendees,” Mesulam says, adding, “The Neurobehavioral Clinic is our principal venue for transmitting the benefits of all developments in the field to our patients and families — and, in turn, patients who volunteer for research allow us to make new discoveries on the cognitive neuroscience of memory, language and behavior.”

Starting at Synapses
For scientists like Savas, the biggest Alzheimer’s breakthroughs will come from investigating the origins of the disease.
“It’s like if a car breaks down: If the battery’s no good, you can’t turn on the radio,” he says. “We want to know what happens first, before it all starts to fall apart.”
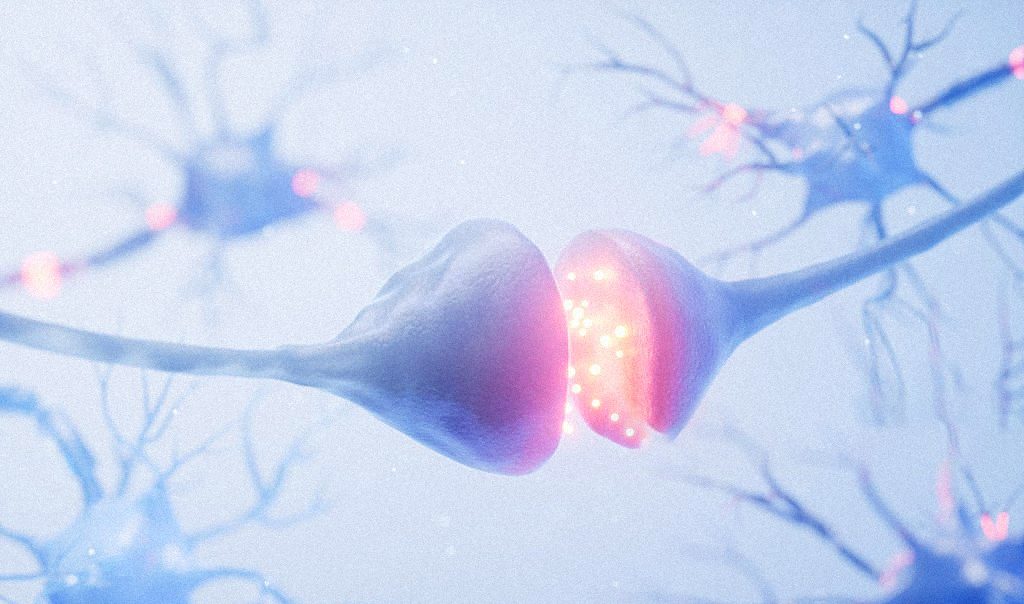
Which is why Savas is currently focused on synapses, the structures that let neurons “talk” to one another.
“Synaptic dysfunction happens very early in the pathology, long before the brain cells die,” he says.
Mutated amyloid proteins play a part here, causing detrimental effects on synapse function, impairing synaptic transmission and promoting neurodegeneration. According to Savas, investigators typically believed that the post-synaptic terminal — the receiving end — was where the primary pathology was, but in a 2017 study published in Cell Reports, Savas found something different.
“We found the presynaptic terminal is actually the first place where it starts,” Savas says.
This lines up with another recent discovery: Last year, Savas worked with scientists across the world to uncover the normal, non-mutated function of amyloid protein. Using a biochemical interactions screen designed by Savas, the investigators discovered that amyloid proteins normally bind to an inhibitory neurotransmitter receptor — again, in the presynaptic terminal.
These findings, published in Science, suggest that the presynaptic terminal may be where Alzheimer’s starts — or at least a promising therapeutic target, Savas says.
“We think the presynaptic terminal represents an underappreciated location for amyloid protein toxicity,” Savas says. “It tells us a small but important piece of the puzzle.”

Other scientists are also examining synaptic regulation’s role in Alzheimer’s disease, including Peter Penzes, PhD, the Ruth and Evelyn Dunbar Professor of Psychiatry and Behavioral Sciences. In a study published in Molecular Psychiatry, Penzes found that Alzheimer’s-associated mutations in a gene called BIN1 can disrupt synapse-synapse communication. While it might not be enough to cause Alzheimer’s disease on its own, Penzes believes BIN1 could be a contributor.
“If a patient has one mechanism of disease, like this gradual weakening of synapses, they might be more sensitive to other mechanisms, like amyloid protein buildup,” says Penzes, also director of the Center for Autism and Neurodevelopment and a professor of Physiology.
Still Fighting the Good Fight
Despite a new emphasis on understanding the root cause of disruptions in synapse-to-synapse communication, investigators aren’t giving up on fighting amyloid proteins.

Recently, Robert Vassar, PhD, the Davee Professor of Alzheimer Research and professor in The Ken and Ruth Davee Department of Neurology, was involved in a study that discovered a treatment originally developed to help patients recover from stroke may also have beneficial effects for Alzheimer’s disease. The drug, 3K3A-activated protein C (3-APC), significantly slowed production of harmful proteins in mouse models of Alzheimer’s by inhibiting a gene called BACE1, according to the study published in the Journal of Experimental Medicine.
This had downstream effects: Mice modeling Alzheimer’s treated with 3-APC performed better on cognitive testing when compared to mice who received the placebo treatment. In fact, they tested nearly as well as healthy mice, demonstrating that reducing amyloid buildup could be effective in ameliorating cognitive symptoms.
Repurposing the stroke drug 3K3A-APC for Alzheimer’s disease is an exciting possibility, given the paucity of treatments for this devastating neurodegenerative disease.
Robert Vassar, PhD
“Repurposing the stroke drug 3K3A-APC for Alzheimer’s disease is an exciting possibility, given the paucity of treatments for this devastating neurodegenerative disease,” says Vassar, who is also scientific director of the Division of Behavioral Neurology, a professor of Cell and Developmental Biology and director of the Alzheimer’s Disease Core Center grant.
However, inhibiting BACE1 has its downsides: In a different study, published in Science Translational Medicine, Vassar discovered that another promising BACE1-inhibiting drug had the potential to cause new defects in the hippocampus.
The investigators discovered adult mice exposed to BACE1 inhibition had deformations in the mossy fiber pathway, a structure in the hippocampus important for memory. With several clinical trials of BACE1 inhibitors either in progress or on the horizon, this is concerning, according to Vassar.
“You don’t want to create a new disease when you are trying to cure another one,” he says.
But there’s still hope. Vassar believes there might be a sweet spot of inhibition: Low enough to keep the mossy fiber pathway intact, while still high enough to significantly decrease amyloid protein production.
“I want to try and find out at what level of inhibition this occurs,” Vassar says. “I think if you can fine-tune it and start early enough, you can have a huge impact.”
Clinical Care and Beyond
All this science leads to one place: the clinic, and Feinberg clinicians are improving care there, too. One recent study found a new, noninvasive way to diagnose early cognitive impairment, the earliest stage of Alzheimer’s disease where affected individuals become forgetful, but still can maintain independence.
Amani Fawzi, MD, the Cyrus Tang and Lee Jampol Professor of Ophthalmology, led the study, published in PLOS One, which found patients with mild cognitive impairment had fewer blood capillaries in the back of the eye, detected using a simple infrared camera.
“Once our results are validated, this approach could potentially provide an additional type of biomarker to identify individuals at high risk of progressing to Alzheimer’s,” says Fawzi. “These individuals can then be followed more closely and could be prime candidates for new therapies aimed at slowing down the progression of the disease or preventing the onset of the dementia associated with Alzheimer’s.”
Science feeding directly into clinical care is a core pillar of the Mesulam Center’s patient care mission, and will continue to fuel future growth, according to Mesulam.
“We will learn more about the neurocognitive networks that enable us to find the right word and recall recent experiences; we will learn more about the mechanisms of selective vulnerability in neurodegenerations; and we will learn more about the reasons why a specific disease causes a specific set of impairments in individual patients,” Mesulam says. “Through these interactive developments, the clinical arm of the Mesulam Center will be able to practice its own brand of personalized precision medicine.”
Above all, the Mesulam Center is well positioned to embark on this new phase of Alzheimer’s research: pulling from all areas of science and medicine to create a new, integrative model of Alzheimer’s disease, in order to stop it in its tracks.